




站在2021年當下,NGS技術已投入使用十餘年,從科技殿堂拾階而下,應用到臨床和社區抗疫一線,回顧底層技術發展脈絡,溫故知新。短讀長、長讀長各有哪些優勢?基礎原理是什麼?主要廠家有哪些?大阪大學微生物病研究所感染症國際研究中心特聘副教授中村昇太(Nakamura Shota)在《生物工學會誌》期刊上發文闡述了相關問題,本文編譯原稿如下。
撰文/中村昇太 編輯/基因慧July 審核/Barney
【注】本文是作者於2020年執筆,發表於《生物工學會誌》2021年5月25日,旨在記錄當下NGS現狀;以下內容編譯原文,略有刪減,僅做科普和研究參考)
自2005年下一代定序技術(Next Generation Sequencing,NGS)問世以來,不斷演變出各種類型,興衰交替。
例如,來自454 Life Sciences公司的焦磷酸定序法,是世界上首個進入市場的下一代定序技術,但如今公司和技術都已鮮有身影。此外,還有一些技術只被部分機構引進和使用,而有些技術大肆宣稱著來年發佈但最終未能問世,這樣的情況不在少數。
定序領域出現了如此多的技術消亡算是十分罕見。而能夠在這場技術競爭中勝出的技術,如今已成為生命科學不可或缺的工具之一,頻繁出現在知名的學術期刊中。
在名稱方面,目前普遍認為“下一代定序技術”這一術語已然不合適,也有稱該技術為大規模平行定序技術(Massively-Parallel Sequencing,MPS)或高通量定序技術(High-Throughput Sequencing,HTS),本文仍採用NGS的說法,是希望將它作為改變基因某個時代的象徵符號來使用,我們知道,在NGS技術出現後,組學領域的定序方法發生了巨大革新。
短讀長定序技術發展
454焦磷酸定序技術是初代的NGS技術,以454 Genome Sequencer 20 (GS20)這一型號定序儀上市,序列讀長平均為100bp(bp:堿基長度),序列數據量約為50Mb(Mb:100萬堿基)。
其後,Roche收購了454 Life Sciences繼續進行開發工作,但在2016年宣佈退出市場。大阪大學微生物病研究所的附屬機構遺傳信息實驗中心首次引進的NGS儀器是就是桌面型小型儀器454GSJr。該儀器於2010年經引進,距其退出市場僅短短6年。
2006年左右,GenomeAnalyzer (GA)問世,該機型採用了由Solexa公司開發的Sequence-By-Synthesis (SBS)法,該技術首次實現了Gb規模的資料量。Solexa後被Illumina所收購,GA通過HiSeq系列得以傳承下來。
另外,善用桑格法的Applied Biosystems(ABI)公司也研發出了Sequence By Oligo Ligation and Detection(SOLiD)技術,當時Roche、Illumina、ABI三巨頭之間展開了激烈的研發競爭。而後,ABI成為了Life Technologies公司的一部分,投入使用了Ion PGM和Ion Proton 等新型半導體定序技術。
經過幾番激烈的技術競爭,目前Illumina公司推出的SBS方法最為廣泛使用。上述兩種技術的解讀序列長度都比現有的桑格定序法短,因此也被稱作短讀長定序技術。以桑格定序法為第一代,這些短讀長定序技術被稱作第二代定序技術。
第二代序列通過在微觀規模空間內進行PCR反應來放大1分子的DNA,基於大規模平行的反應產生巨大的資料量,因此被稱作大規模平行定序技術。這種大規模平行性逐年提高。Illumina最新機型NovaSeq6000實現了Tb級別的資料量。在這15年間產生了10的6次方的巨大變化。
當454焦磷酸定序技術問世並隨之推出其小型機454GSJr時,有評價稱可以通過該項技術實現基因解讀的平民化。也就是說,過去只有部分大型機器才能完成的基因組解析,如今一般的研究室也可以完成了。
相比而言,NovaSeq進行人類全外顯子組分析時,一次定序運行可分析200個樣本,對於用途廣泛的RNA-Seq則可分析400個樣本,僅用兩天左右的時間就能輸出資料,這對於單個研究室的需要來看已然不可能,以部局、大學為單位也十分困難。為此,遺傳信息實驗中心正在廣泛招募國內外用戶來推進機器的共同使用。最近,不僅是學術機構,普通企業也願接受邀請來促進產學合作,但目前仍是為滿足一台NovaSeq的需求而精疲力盡的狀況。
另外,Broad Institute、Sanger Institute、BGI等大型分析中心擁有達到了數十PB規模存儲系統的大型電腦設備,並使用多台NovaSeq,每天都能生產出大組學資料。基因組解析再次復興時代,被解析的樣本數也計畫達到數十萬規模。事實上,計畫在荷蘭進行的一項名為lifelines的專案中,預計通過宏基因組鳥槍法對10萬人的腸道細菌進行大規模群體研究分析。在論文中已經成千成萬關於「Population-based」的研究,可以預見到今後基因研究資料將愈發大型化。希望在日本也建立起能夠與世界大型分析中心抗衡的設施。
長讀長定序技術的發展
相較於此前所闡述的短讀長定序技術,2010年左右,Pacific Biosciences公司開發的Single Molecule Real Time Sequencing(SMRT)技術在解讀序列長度上實現了飛躍性的延伸進步。從該技術問世之初,就因聲稱可以超越桑格定序法的讀長達到數kb而震驚全球。
2013年左右,PacBio RSII經發佈,實質上的廣泛利用就是從此時開始的。該技術基於即時監測1分子聚合酶的延伸反應的原理,由於與第二代微型PCR為基礎的原理具有革新性的不同,因此也被稱作是第三代定序技術。
當前SMRT技術的最新機型Sequel II的性能具有顯著的提高,每SMRT1細胞單位為150Gb,平均讀長分佈約為50kb。上市當時該機型在解讀序列的準確性較低,被視作一大問題。通過使用數次重複解讀環狀化DNA庫的Circular Consensus Sequencing(CCS)的方法後,一個分子由來可達99%以上準確度的高精度讀長(HiFi讀長)得以實現,通過由數個Subreads序列(CLR)形成共有序列的方法,據說可以達到99.999%以上的準確度。
隨著SMRT技術的出現,de novo assembly(無參照序列的基因組解析)領域最先產生了革新性變化。雖然隨著第二代定序技術的發展各種基因解析方面取得了一定進展,但即便是微小的微生物基因組,也不可能完全獲得長基因組序列。因為短讀長的數百個堿基長度無法完全確定基因組中多次重複出現的核糖體排列等位置關係。通過SMRT技術長讀長定序才得超越核糖體排列全長、並根據核糖體兩端的序列資訊確定絕對位置(圖1,參考文獻1)。
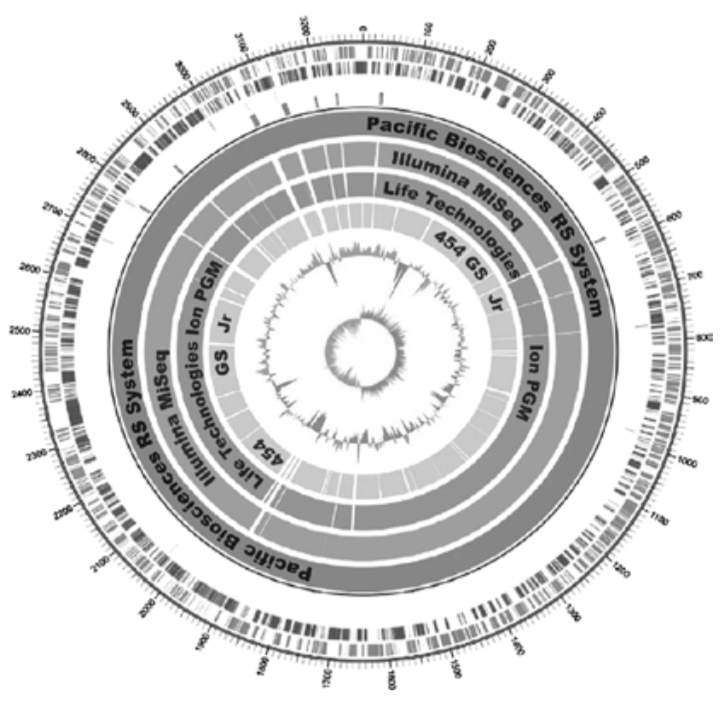
圖1:利用第二代和第三代定序技術對副溶血弧菌腸炎基因組進行基因組分析比較。從內側開始,是通過454焦磷酸定序、Ion PGM、Illumina、PacBio各項技術得出的重疊群序列在參考基因組上的結果。環狀的缺口是無法解讀的地方。缺口與從外側往裡第4個所示的rRNA的位置一致。根據PacBio的分析結果,一條重疊群變成了染色體(來源/生物工學會誌)
另外,Oxford Nanopore Technologies(ONT)公司研發的Nanopore定序技術也屬於長讀長定序技術。該技術通過DNA長鏈分子穿過由細菌毒素產生的膜蛋白質的細孔結構(Nanopore)時的離子電流的變化來解析核酸的種類。
桑格法和NGS技術從來都是依賴于聚合酶的延伸反應,而通過該技術得以不依賴聚合酶就能獲得堿基序列資訊,因此可以將其稱作是第四代技術。它的最長讀長超過4 Mb,似乎只要DNA品質足夠好,無論多大都能進行解析。因此,如何製備長DNA分子這一有關DNA提取的問題就變得至關重要。
這項技術的一大特點是設備的小型化。最初發佈的MinION只需手掌大小的尺寸就能進行細菌基因組解析,因此全世界的研究人員可以將其攜帶外出,隨之就是否可將設備攜帶至原始森林、山中等極端困難的場所進行定序而展開了激烈角逐。現在已經發展到可以攜帶至宇宙空間站。
如今,不僅是MinION,大型機器PromethION和逐步小型化的Flongle等新設備不斷推出。如上所述,ONT公司的研發能力十分驚人,Basecaller(變換序列的軟體)每隔幾個月就進行更新,解析精度也實現了日新月異的改善。現在的MinION輸出的原始資料的速度可達到每小時約20GB。換算成每分鐘約為330MB /min,與4K視頻(350Mb /min))的信息量相當。雖然外觀小巧,但處理其輸出的大資料還需大型電腦才行。
筆者希望利用這種體積小且解析速度快的優點,將其應用於病原體的當場識別。這種小型裝置也使得設置在醫院的檢查室成為可能。但是,由於配備大型電腦比較困難,所以需將解讀後的序列資訊發送到遠距離的大型電腦,由該電腦進行高級分析,然後將結果回饋至使用者終端(圖2)。實際上,有關通過雲計算識別病原體的應用,以非結核性分枝桿菌病(NTM)為例開始實驗,在醫院的檢查室裡10分鐘的定序時間中即時同步進行堿基序列解析,便可以甄別菌種(參考文獻2)。
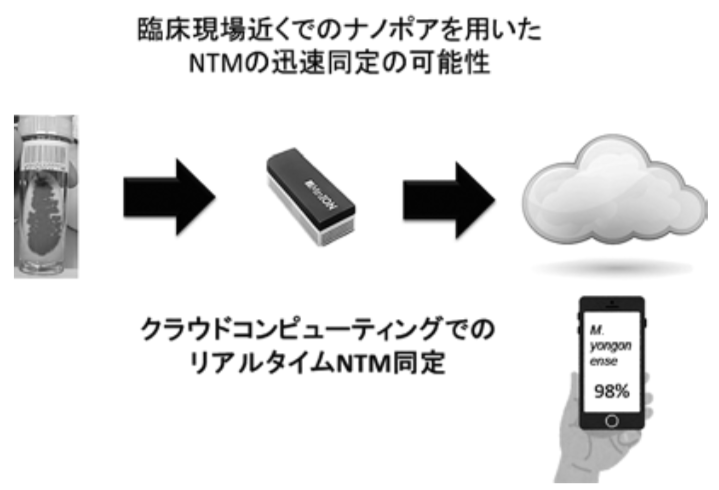
圖2:開發中的非結核性分枝桿菌病(NTM)的迅速當場識別法。從檢查室培養的分岐桿菌中提取DNA,在檢查室實施Nanopore定序,通過雲端即時分析(來源/生物工學會誌)
新一代定序技術
近年來,華大集團旗下的子公司華大智造所開發的DNBSEQ定序技術發展迅猛。這項技術原是由於2006年成立的美國Complete Genomics公司所開發。華大集團於2013年收購了該公司,此後結合自身技術逐漸發展壯大。
其原理屬於第二代定序技術,通過將環狀化的1分子DNA範本放大到被稱作「DNA納米球 (DNB)」的球狀的DNA高分子中進行解析。
目前,日本已經引進了多台華大智造MGISEQ-2000RS基因定序儀。值得一提的是,該機器擁有Illumina公司尚未實現的技術,可實現以往以300bp為極限的單端400bp序列的定序模式。此外,華大智造還推出了基因定序儀DNBSEQ-T7,在長期以來Illumina公司獨佔鰲頭的短讀長業界,華大智造儼然具有遊戲顛覆者的趨勢。
最近華大智造還開發了名為CoolMPS的新型核酸視覺化技術並就其發表了論文(參考文獻3)。該技術通過能夠識別天然核酸的核酸特異性抗體來解析核酸的種類,因此具有在聚合酶的延伸反應中不易產生錯誤的特點。該配套元件也已經發佈,通過CoolMPS所獲的資料也正被廣泛使用中。華大智造在今後還預計推出單細胞分析用的小型設備以及更加先進的產品,值得持續關注。
展望未來

圖3:現有主力機種,從左至右分別為NovaSeq6000,Flongle,MinION,PromethION,MGISEQ-2000,DNBSEQ-T7,PacBio-Sequel II(來源/生物工學會誌)。
上述內容介紹了下一代定序技術的歷史以及現有主力機型(圖3)的原理和性能。
NGS所具備能力的主要特徵在於產出的資料量及能夠解析的讀長,其中NovaSeq和T7在資料量方面表現突出;而在讀長方面,Nanopore則具有根據其製備的長鏈DNA進一步延長讀長的潛力。換而言之,它們都已達到了最高性能,至於今後的目標性能,則是精度和資料量達到NovaSeq水準,以及與Nanopore水準相當的長讀長的小型設備。
當這樣的設備出現時,我們還能夠熟練使用嗎?目前,新技術的開發仍在繼續,採用固體材料的細孔結構而非生物聚合物的第五代Nanopore定序技術正在誕生(參考文獻4和5)。而今後對NGS輸出的資訊進行分析的能力,以及支撐其分析的電腦基礎將變得十分重要。
參考資料:
1) Miyamoto, M. et al.: BMC Genomics, 15, 699 (2014).
2) Matsumoto, Y. et al.: Emerg. Microbes Infect., 8, 1043 (2019).
3) Hahn, O. et al.: Nucleic Acids Res., 49, e11 (2021).
4) Di Ventra, M. and Taniguchi, M.: Nat. Nanotechnol., 11, 117 (2016). 5) Goto, Y. et al.: J. Hum. Genet., 65, 69 (2020).
原文出處:基因慧
【聲明】為了服務基因及數位生命健康產業發展、科技推廣及政產學研用連接,本刊中國大陸策略夥伴──基因慧秉持專業、賦能、中立的理念收集、分析、發佈資訊或見解。但由於時效性及行業特殊性,所刊登內容僅供研究參考,不作為決策依據;本文相關資訊不代表基因慧的觀點;基因慧平臺刊登的原創內容的智慧財產權為「基因慧」商標擁有者及相關權利人所有;歡迎轉載,轉載請申請並注明來源。歡迎個人及機構投稿及合作。



