




(編按:本草案於2024年10月時更新,將原先考慮將日本醫藥品醫療機器綜合機構(Pharmaceuticals and Medical Devices Agency, PMDA)納入CRCs清單的構想刪除。)
一、緣起
英國於2020年脫歐後,歐盟的醫療器材法規在英國已不再適用,故MHRA也預計最遲於2030年6月30日前的過渡期前暫時認可歐盟所認證的CE標示,且承認其他具指標性之監管機構所認證的醫療器材,減少在監管機構間進行重複性的評估,並可更快速地進行上市批准,借此確保國內的醫療器材進口不致匱乏。二、預期施行方法
1.英國政府預計直接承認來自參考監管單位所批准的醫療器材,並已將參考監管機構(comparable regulator countries, CRCs)的清單納入本次的政策框架中,CRCs清單包含美國食品及藥物管理局(Food and Drug Administration, FDA)、加拿大衛生部(Health Canada)、歐盟/歐盟經濟區各成員國之主管機關,以及澳大利亞醫療產品管理局(Therapeutic Goods Administration , TGA)。2.除此之外,該產品還需遵循幾項具體規範,如需英文包裝及標籤、產品識別碼、遵循英國對醫療器材產品的電子相容性、貼標材料、上市後監督等管理要求。
3.未來將以提供認可證書的方式,授予醫材進入英國市場的資格,並非使其直接取得UKCA的標誌或認證。
4.本次提出的政策框架仍是草案,目標是2025年開始能夠正式施行最終版本。
三、排除適用的產品
1.經豁免的非商用器材(In-house Device)2.客訂醫材 (Custom-made Devices)
3.不符合英國預期用途指引的醫療器材軟體(Software as a medical device, SaMD)與人工智慧軟體醫材(AI as a medical device, AIaMD)產品
4.早先即透過類似的認可途徑,進入到以上參考監管機構市場的產品
5.透過實質等同性比對途徑(即美國510(k)程序)所批准的SaMD與AIaMD產品。
6.透過實質等同性比對途徑所批准的第IIb等級的植入式醫材與第 III等級醫材。
7.透過實質等同性比對途徑所批准的伴隨式診斷器材。
8.含有「未於英國獲得許可之藥物」的伴隨式診斷醫材與複合醫材。
9.在英國 MDR 2002 Regulation 3列表以外的產品。
四、成為認可醫材的程序
1.自助註冊以下類別的產品如果已透過四個CRCs核准,可直接進行「自助註冊」程序獲得認可資格:
■第I等級醫材,但排除Is(滅菌類)、Im(測量類)與 Ir(重複使用類)。
■非為滅菌類之第A等級體外診斷醫材(In Vitro Diagnostic Devices, IVD)。
以上產品需符合以下列要求:
製造商需進行符合品質管理系統的聲明(如ISO 13485或其他等效標準)。
2.申請提交
以下符合歐盟MDR的醫材,但排除 AIaMD及產品分類在CRCs與英國之間不同之產品:
■符合EU MDR 的Is/m/r等級醫材。
■符合EU MDR的IIa、IIb、III 等級醫材。
■符合EU IVDR的滅菌類第A等級IVD 。
■符合EU IVDR的第B、C、D等級IVD。
以上產品需符合以下列要求:
製造商需提供相關佐證文件包含上市後監督計畫,以及上市後監督報告或定期安全更新報告。
3.附帶特定要求的申請提交
包含以下符合澳大利亞及美國上市核准規範的醫材,亦排除 AIaMD及產品分類在CRCs與英國之間不同的產品:
■第Is/m/r 等級醫療器材。
■第IIa、IIb、III等級醫療器材。
以上產品需符合以下列要求:
「2. 提交申請」所載之全部要求。
製造商需為植入式醫材額外提供植入卡(Implant cards)資訊。
需對第III等級醫材以及植入式醫材進行產品安全性和臨床效能之總結說明。
4.附帶特定要求,且需經過簡要評估的申請提交
包含以下類別的產品:
■符合加拿大醫材法規或美國De Novo、510(k)之第Is/m/r 等級醫材。
■符合加拿大醫材法規或美國 De Novo之第IIa、IIb、III等級醫材。
■符合美國510(k)之第IIa、IIb(非植入式)和 IIb(具成熟技術)等級醫材。
■符合澳大利亞、加拿大醫材法規或美國PMA、De Novo或510(k)的滅菌類第A等級IVD 。
■符合澳大利亞、加拿大醫材法規或美國PMA、De Novo 或510(k)的第B、C、D等級IVD 。
■符合澳大利亞、加拿大、歐盟的醫材法規或美國PMA、De Novo的AIaMD 。
■CRC中的分類與英國醫療器材法規的分類不同的任何醫材 。
以上產品需符合以下列要求:
製造商須符合「2.提交申請」所載之全部要求。
製造商需為植入式器材提供植入卡和病人資訊。
須對第III等級、植入式醫材以及第C和D等級之IVD,進行安全性和臨床效能之總結說明。
須對 AIaMD產品審查上市前訓練及測試資料、效能與確效驗證的實施方式及預訂變更控制計劃。
由於此途徑包含CRC之分類與英國醫療器材法規中的分類不同的器材,因此需要對這些醫材的分級先進行審查。
圖一 英國醫材國際認可(International recognition)申請路徑
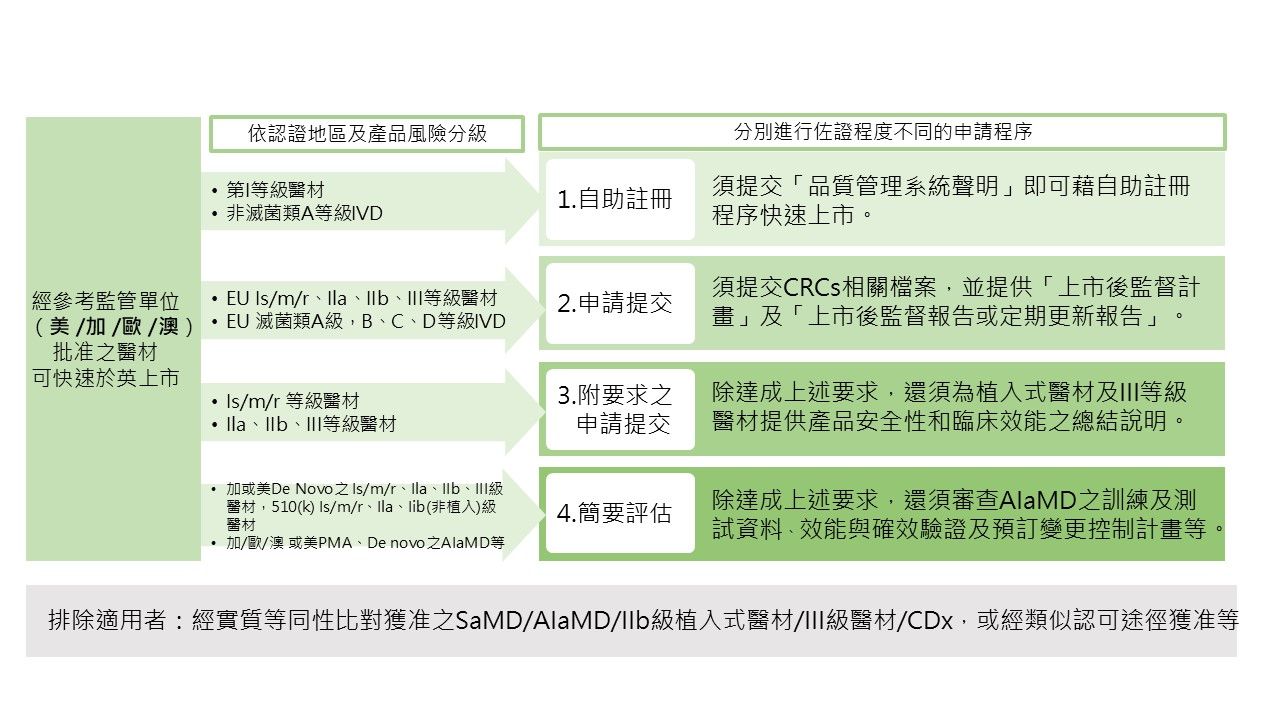
(資料來源:資策會科法所提供)
五、小結
此政策透過值得信賴的監管單位把關,不僅可在英國脫歐後的過渡期間促進醫材之貿易流通,更可能有效減少英國當局於審查過程的行政成本,進而提升國內的產品審查效率。然值得注意的是,雖然英國針對不同地區及不同產品風險等級建立了四條不同審查門檻的認可路徑,但在各國醫療器材監管法規與行政裁量基準不完全一致的現況下,各國政府對於臨床資料及健康風險的解釋與判斷結果也不見得相同,未來在醫療器材上市審核及上市後監督等過程中,將如何看待及利用來自CRCs之證明文件,有待未來持續觀察其實施成效。
撰文/資策會科技法律研究所 劉怡 副法律研究員
資料來源:
1. MEDICINES AND HEALTHCARE PRODUCTS REGULATORY AGNECY [MHRA], MHRA announces a proposed framework for international recognition of medical devices (2024), https://www.gov.uk/government/news/mhra-announces-a-proposed-framework-for-international-recognition-of-medical-devices (last visited Oct. 29, 2024).
2. MHRA, Statement of policy intent: international recognition of medical devices (2024), https://www.gov.uk/government/publications/implementation-of-the-future-regulation-of-medical-devices/statement-of-policy-intent-international-recognition-of-medical-devices (last visited Oct. 29, 2024).



