




2022 年全球罕病治療市場為1,284 億美元,預計2030 年達到3,3584 億美元,複合年成長率高達12.8%!
臺灣《罕病法》上路比歐盟更早,居亞洲第一國,不論是對孤兒藥的鼓勵誘因、對罕病族群的關懷心志,將屆滿23 年來,這部大法大力支撐著國內患者的權益。
盤點國內罕藥/ 孤兒藥研發公司,有超過30 間之多,回顧2022 年,臺灣新藥/505(b)(2) 證取證突破10 張,創下產業紀錄,其中,罕病/ 孤兒藥佔最大宗,盤據臺灣新藥研發領域。
對過去無藥可醫的患者而言,這群生技公司,宛如黑暗中的曙光。但下一個20 年間,臺灣還能持續站在世界罕病大國的行列中嗎?
專題企畫:巫芝岳
撰文、攝影:巫芝岳、李林璦
採訪整理:巫芝岳、李林璦
視覺/美術設計:黃黛鵑
特別感謝:華淵鑑價股份有限公司提供報告
參考資料:市場公開資訊
圖:Vecteezy, freepik
這一、兩年來,疫情最嚴峻期間,臺灣多間罕病藥物開發公司,臨床成果卻紛紛傳出捷報!
2022年12月11日,漢達(6620)美國子公司Handa Neuroscience的多發性硬化症505(b)(2)新藥Tascenso (0.5 mg),取得美國食品藥物管理局(FDA)的最終核准,股價也一路上飆,闖入百元俱樂部。
專攻罕見眼科病變斯特格氏症(Stargardt Disease)新藥的仁新醫藥(6696),子公司Belite Bio去年4月正式於那斯達克(Nasdaq)掛牌上市,當時IPO籌集了3,600萬美元,股票價值飆升超過500%,市值也在7月時突破10億美元,成為臺灣近年來唯一一家新藥研發新創獨角獸。
此外,2022年2月登興櫃的國邑(6875),聚焦原發性肺動脈高壓(PAH) 505(b)(2)新藥;以及2021年11月,藥華藥(6446)以國外罕病真性紅血球增生症(PV)藥物BESREMi取證,一度帶動國內生技股價全面上漲等,皆是以罕見疾病為標的的新藥公司。
在寒冬來襲的資本市場中,這幾家生技新藥公司的逆勢,支撐了許多產業分析師口中「生技股抗跌」的預言。
這些選擇以「小眾」的罕病市場為題的生技公司,紛紛異軍突起,不僅成為臺灣生技醫藥類股的新尖兵,也讓臺灣躋身罕藥/孤兒藥研發國際列強之列。

漢達美國子公司Handa Neuroscience 的多發性硬化症505(b)(2) 新藥 Tascenso (0.5 mg),取得美國FDA最終核准。圖為漢達總經理陳俊良。( 攝影/ 羅翊方)。
孤兒藥鼓勵誘因強 FDA半數批准藥物獲ODD
根據市調公司Grand View Research調查,2022年全球罕病治療市場為1,284億美元,預計2030年可達到3,358億美元,複合年成長率(CAGR)為12.8%。
根據華淵鑑價近期統計,2018~2021年FDA批准的孤兒藥(Orphan Drug)項目連年創下新高,從2018年的68項,增加到2021年的132項,四年間成長了將近兩倍。
2021年,FDA所核准的50項新分子實體(New Molecular Entities, NME)藥物中,就有26項為孤兒藥。
FDA為促進罕見疾病療法開發,祭出了孤兒藥的種種鼓勵措施,包括取得「孤兒藥資格」(Orphan Drug Designation, ODD)後,臨床試驗支出可抵稅、獲批上市後擁有7年獨賣權等,不但促使更多廠家投入研發,也讓許多過去無藥可醫的罕見疾病患者,看到了曙光。
對於新藥開發資源有限的臺灣而言,在法規與行政流程的優惠下,孤兒藥儼然成為國內生技公司開發的重要策略選項之一。
各國「罕見疾病」定義不相同 臺灣納入「疾病須有遺傳因素關聯」
不過,全球各國對「罕見疾病」定義不盡相同,美國所定義的罕病,為每年新病患人數少於20萬的疾病,針對此些疾病的藥物可列為孤兒藥;而臺灣則定義為患病人數在萬分之一以下。
此外,根據衛福部的「罕見疾病審議認定原則」,除了盛行率需在萬分之一以下外,考量疾病嚴重度,並排除人為外在因素所造成之疾病或傷害,以及癌症外,也特別將「疾病須有遺傳因素關聯」列入。
本刊採訪的多位專家,包括:神經罕病專家彰化基督教醫院副院長劉青山、中華民國罕見疾病製藥發展協會秘書長朱璿尹、罕見疾病基金會創辦人陳莉茵等皆指出,臺灣對罕病定義相對嚴格,主要是與我們的藥品牽涉「罕見疾病藥物」的認定,以及健保給付等財務因素相關。
全球首部「罕病防治+罕藥」法規利基

國健署署長吳昭軍說明,該法規立法目地包括防治、及早診斷、照顧、協助取得藥物,及鼓勵相關藥物和特殊營養食品的製造和研發。( 攝影/ 巫芝岳)
臺灣在2000年通過《罕見疾病防治及藥物法》,奠定了至今20多年來,臺灣的罕病醫藥基礎。
《罕見疾病防治及藥物法》是造就臺灣一度為全球「罕病大國」的一大因素,不但使病人權益受到保障,對國外藥商而言,也是吸引其進入臺灣的一大誘因。
當年,在罕病基金會及許多產、官、學界人士的積極參與下,促成了該法的立定,也讓臺灣成為繼美國、日本、澳洲、歐盟後(編按:發佈時間甚至比歐盟早一天),全球第5個立法保障罕病病人的國家,該法的內容,同時結合罕病防治與罕藥法,是世界首見同時保障防治與藥物的罕病相關法規。
國健署署長吳昭軍,於去(2022)年10月1日臺大藥學專業學院主辦「罕見疾病用藥可近性論壇」中說明,該法規立法目的包括防治、及早診斷、照顧、協助取得藥物,及鼓勵相關藥物和特殊營養食品的製造和研發。
該法主管單位涵蓋健保署、國健署和食藥署,國健署主要負責罕病法規、防治工作補助、宣導教育等;食藥署主管罕藥法規;健保署則負責患者就醫醫療費用、核發重大傷病證明等。
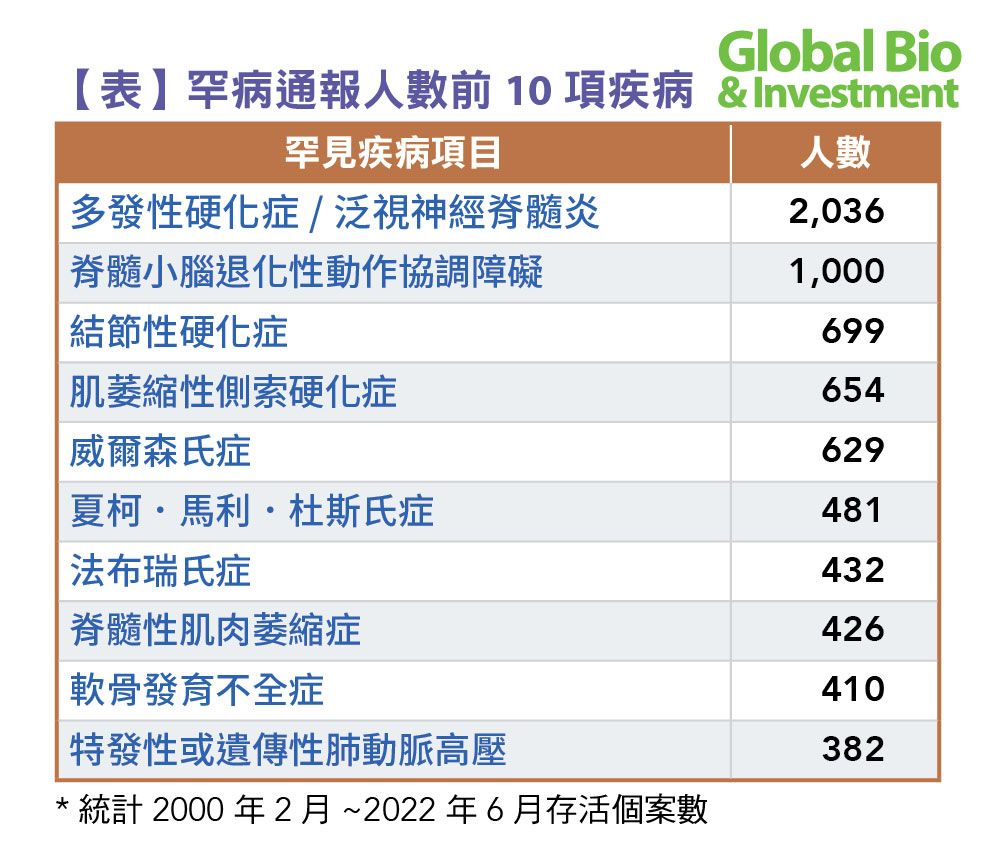
從2000年2月法規通過後,目前,國內認定的罕見疾病共有240項;至2022年11月30日,通報案數為19,699人,其中以多發性硬化症(MS)/泛視神經脊髓炎(NMOSD)為最大宗。
據衛福部統計,2000年2月~ 2022年6月國內患病人數前十名的罕見疾病如下表:
罕藥「快速通道」受外商青睞
在罕病藥物方面,《罕見疾病防治及藥物法》為了促進病人能盡快取得用藥,特別設有「罕藥認定」的機制,藥品須先送至食藥署和罕見疾病及藥物審議會,經審議認定後,若尚未通過查驗登記,得以專案進口/製造管道,若已通過則可申請納入健保。
中華民國罕見疾病製藥發展協會秘書長朱璿尹表示,這項法規相當於只要拿到罕藥認定,就能直接申請健保,當時,過程可能只需約六個月就能順利讓病人透過健保使用到藥物,遠遠較至少一、兩年的藥物取證過程快速。因此,對外商引進罕病藥物來臺灣相當有吸引力。
此外,為了鼓勵罕藥查驗登記,政府也推出多項鼓勵措施,包括:罕藥許可證有效期間為10年,在此期間不受理同類藥物查驗登記申請;取消十大先進國家採用證明之限制;免除樣品送驗,且臨床試驗之數目、受試者人數等方面得視個案考量;查驗登記規費減免(規費約為一般藥品的1/3 )等。
朱璿尹指出,其中「10年專賣期」的設計,更是有利於許多臺灣學名藥廠商,一旦藥品專利過期,就有機會搶先在國外原廠在臺取得罕藥認定和查驗登記,並在臺灣獨賣10年。
不過,朱璿尹也同時表示,該法規其實具有一定的彈性。例如武田(Takeda)與國廠南光化學,幾乎同時提交申請的遺傳性血管性水腫(Hereditary Angioedema)藥物Icatibant,衛福部就都在2021年1月28日給予罕藥藥證,使兩公司同享10年專賣期,不論對外廠或臺廠都有一定的保障。
此外,鑑於國內罕見疾病人數每年約成長8.5%,平均每人藥費約成長17.2%。為了避免罕藥遭到健保預算排擠,如其中血友病人數每年小幅成長2.6%,惟平均每人藥費每年約成長9.4%,醫療費用之成長極為快速。
2005年起,健保就為此規劃了罕病專款「罕見疾病與血友病藥費」預算22.3億新臺幣,且該預算逐年提升,並納入罕病特材。
2022年,健保署這項「罕見疾病、血友病藥費及罕見疾病特材」預算,已達128.37億新臺幣(醫院128.07億新臺幣、西醫基層0.3億新臺幣)。

中華民國罕見疾病製藥發展協會秘書長朱璿尹表示,這項法規對外商引進罕病藥物來臺灣相當有吸引力。( 攝影/ 羅翊方)
臺灣破20家孤兒藥廠 臨床漸開花結果
在法規及健保專款等帶動下,不僅開始吸引外商陸續將藥物送來臺灣申請罕病藥物認定,同時促使國內其他多家製藥公司開始投入罕病及孤兒藥的開發。
國內第一家投入罕病藥物代理的科懋,早在大約1997年,了解到臺灣罕病藥物的缺口後,率先代理多項罕病藥物進入臺灣,包括:法布瑞氏症、肺動脈高壓、紫質症藥物等,並成為《罕見疾病防治及藥物法》的重要推手。
科懋董事長陳澤民表示,科懋成立以來一直鎖定國內仍缺乏的少量、急症藥物進行代理,包括30年前首次引進生長激素藥物、潰瘍性結腸炎藥物,及2005年臺中「毒蠻牛事件」所需的氰化物活性碳解毒劑等。
除了代理外,科懋透過旗下的科進製藥,持續研發包括高血氨症在內的罕病藥物,在2001年取得臺灣食藥署(TFDA)核發的第一張孤兒藥證。
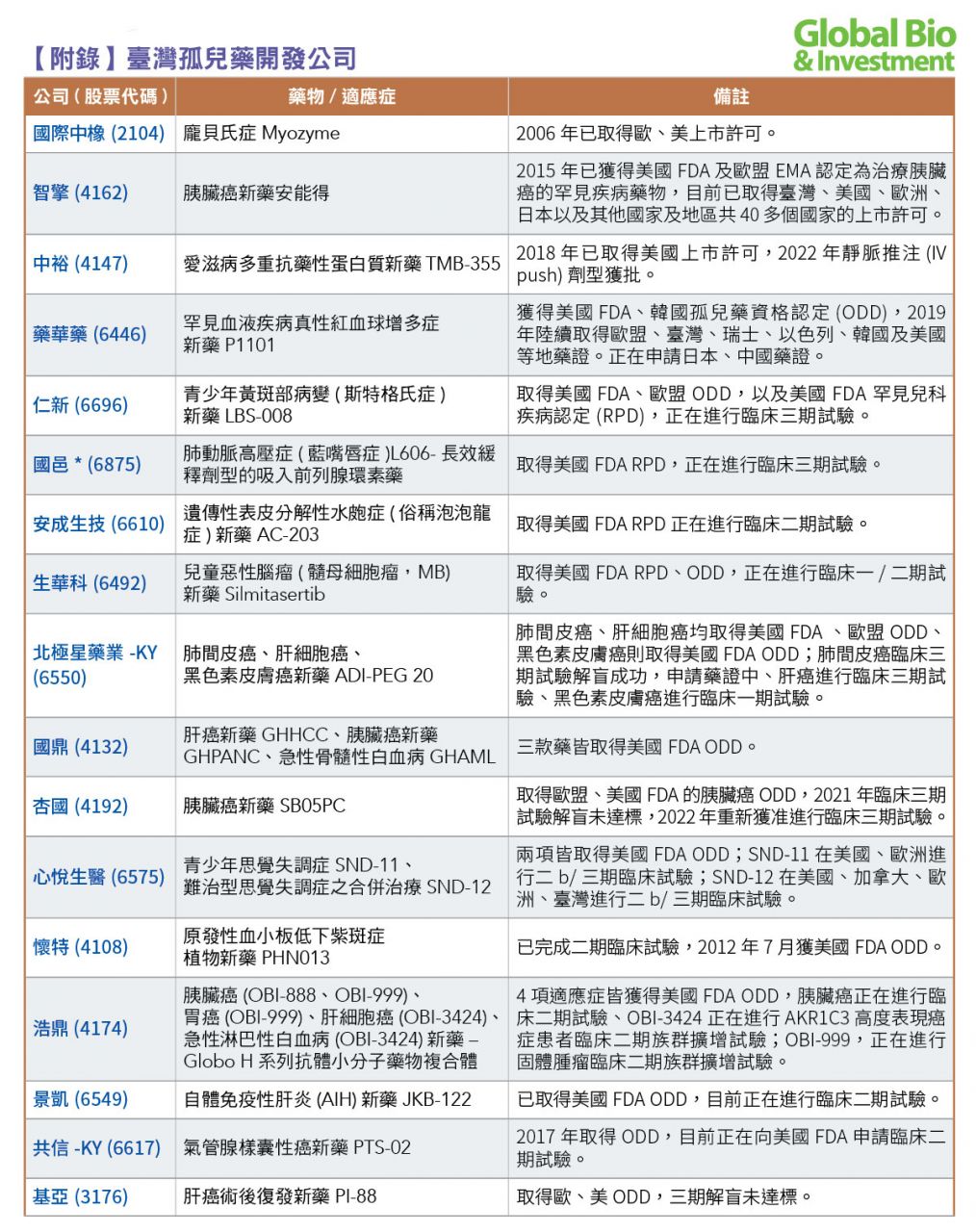
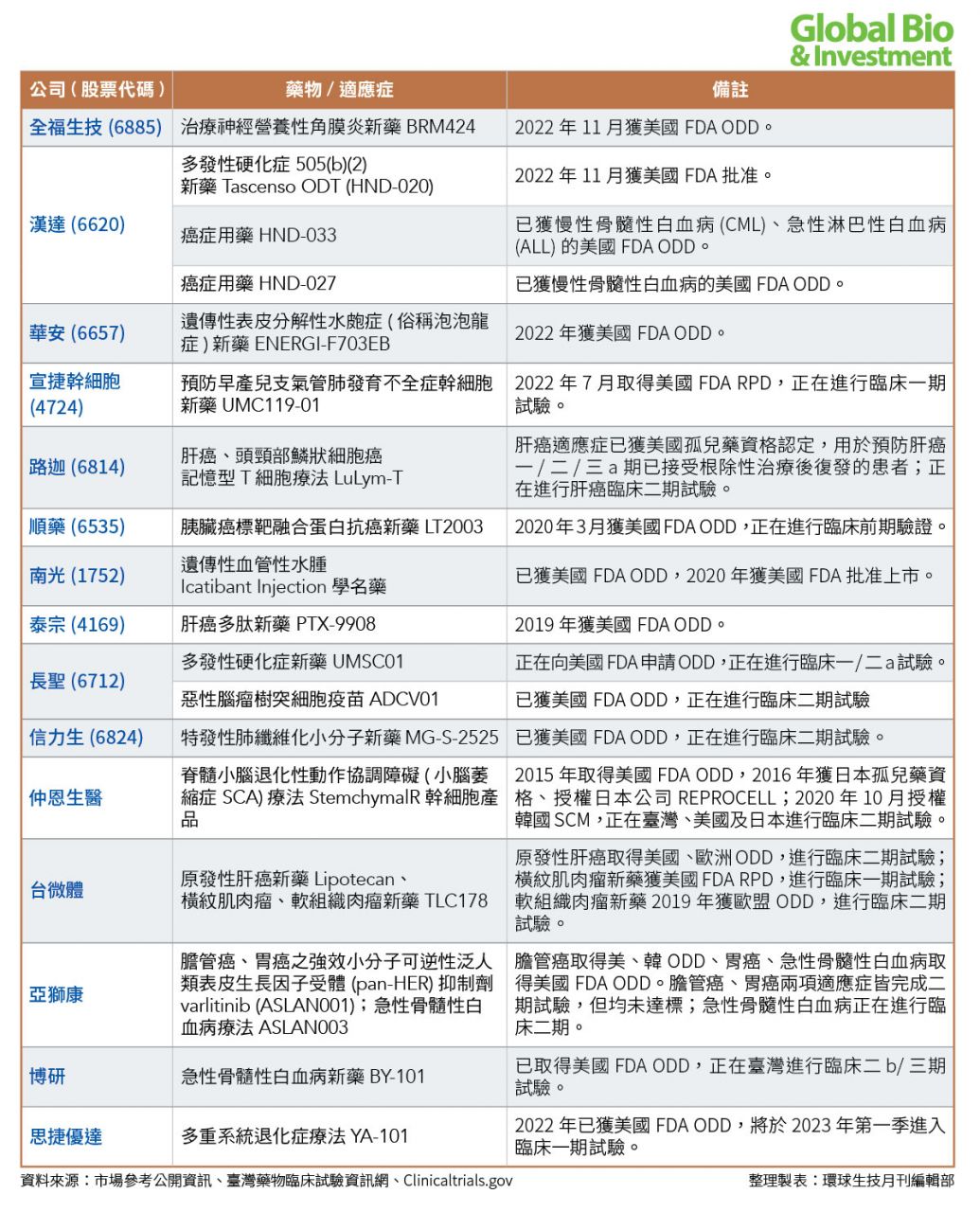
目前,臺灣總計有超過30家藥廠,透過FDA孤兒藥的快速通關方式發展;已有中橡、智擎、中裕、藥華藥、漢達、南光等廠商取證上市。(參見附表)
另外包括:仁新醫藥、安成生技(2022年6月被保瑞併購)、生華科、台微體、浩鼎、北極星藥業、國鼎、杏國、懷特、心悅生醫、順藥、泰宗、共信、華安、全福、思捷優達、博研等26家藥廠旗下藥物,都已獲得美國孤兒藥資格認定。
其中,仁新醫藥、安成生技、生華科、台微體宣捷、國邑,同時也取得罕見兒科疾病認定(RPD),有望加速產品問世。
目前,臺灣有6款孤兒藥品已獲上市。其他大部分孤兒藥新藥雖然處在開發階段,但已有4項走入臨床後期,包括杏國胰臟癌新藥SB05、北極星藥業肝細胞癌新藥ADI-PEG 20、以及心悅旗下難治型思覺失調症之合併療法SND-12也於2021年3月獲臺灣衛福部許可,核准進行二b/三期人體臨床試驗。
多家具潛力的新藥公司,也進入臨床三期階段,包括:仁新醫藥開發的乾性黃斑部病變及斯特格氏症的口服新藥LBS-008,目前已取得澳洲藥物管理局(TGA)、臺灣食藥署獲准,針對斯特格氏症青少年病患進行臨床三期試驗等。
臺大新穎基因療法 技轉外廠2022取證
除了傳統的藥物外,「基因治療」這項新興療法,在經歷十多年突破各種藥物傳輸技術、安全性上的挑戰後,在近十年成為許多難解罕病的重大突破。
自2018年,Spark Therapeutics的Luxturna取得FDA首張基因療法藥證,成為第一款治療遺傳性疾病的基因療法後,全球各廠的研發與取證可說是突飛猛進。
對於許多因遺傳性基因問題導致的罕病,此類得以從根本修正基因的治療方式,突破過往難以「治本」的問題,有些療法甚至能讓病人不需終身用藥,只要治療一次就能痊癒。(全球罕病基因療法介紹,詳見後文)
在臺灣,雖未有本土自產、自行取證的基因療法誕生,但去年7月傳出技轉外廠取證捷報的臺大醫院團隊,可謂本土罕病基因療法的重要開端。
臺大醫院團隊自2007年起,投入開發罕見疾病芳香族L-胺基酸脫羧基酶(AADC)缺乏症基因療法――Upstaza,並於2015年技轉給美國PTC Therapeutics藥廠,2022年7月20日,PTC傳出捷報,此大腦注射基因療法,獲得歐洲藥物管理局(EMA)上市許可,用於治療18個月以上的AADC兒童患者,成為AADC首個療法,也是歐洲首款腦部基因治療藥物。
這項藥物最早是由小兒科教授胡務亮團隊開發,從研發到臨床試驗,都由臺大醫院主導,並在臺灣進行,首位患者已在2010年給藥。
2010年,透過臺大醫院基因醫學部提出「罕見疾病芳香族L-胺基酸脫羧基酶缺乏症治療計畫」後,陸續獲得AADC缺乏症病友會、罕見疾病基金會、長榮集團總裁張榮發、及生技醫藥國家型科技計畫(NRPB)的支持贊助。

2022 年7 月,臺大醫院宣布其技轉給PTC 的罕病基因療法獲歐盟批准。圖左二為領導該技術研發的小兒部教授胡務亮。(攝影/ 彭梓涵)
臺灣新生兒罕病篩檢、防治、早期治療 居世界之首
除治療外,帶動國內罕病藥物發展的另一大動能,在於臺灣有相當完善的篩檢制度,包括:婚前檢查、產前檢查、以及新生兒篩檢。
透過血液、基因檢測,能提早發現下一代可能罹患罕病的風險,無論是在孕前進行防治(如以人工體外受精,選擇健康胚胎著床),或是產前檢查出後進行人工流產; 又或是嬰兒出生後得知,但因及早治療而能提升預後,都是這些篩檢對於罕病防治相當重要的貢獻。
以新生兒篩檢為例,臺灣每位新生兒,一出生滿48小時,就會被從後腳跟採集一滴血進行篩檢,這項制度自1982年起執行。根據罕病基金會資訊,當時僅檢測5項先天性疾病(先天性甲狀腺低能症、苯酮尿症、半乳糖血症、高胱胺酸血症,與俗稱蠶豆症的葡萄糖六磷酸鹽脫氫酶缺乏症);2000年再增加先天性腎上腺增生症;2016年7月指定篩檢項目更增加為11項。
目前,政府針對這11項篩檢項目,補助費用200元、家長自付350元及採檢醫院行政費用或相關材料費。
此外,另有選擇性篩檢的其他選項,例如龐貝氏症(Pompe Disease),就能透過早期篩檢並用藥,明顯改善病童的生活品質,甚至至今,臺灣仍是全球唯一將龐貝氏症納入新生兒篩檢的國家。
在醫界的努力下,罕見疾病篩檢與治療的流程也不斷被改善中。
以龐貝氏症為例,過去標準診斷流程中,新生兒血液檢出疑似龐貝氏症後,需再進行肌肉切片、基因檢測等檢驗,確診後才能向健保申請藥物核可,但走完整個行政流程拿到藥物,幾乎需要3個月的時間,才能開始進行治療。
臺北榮總兒童醫學部主任牛道明領導的團隊認為,由於治療的快慢會直接影響成效,故決定「只要第一次新生兒篩檢達4項診斷標準」,病童就能在到達醫院的4小時內,獲得第一次酵素治療。
至今十多年來,北榮表示,其所有龐貝氏症病童平均都能在9.75天內接受到第一次酵素治療,被全球龐貝氏症專家視為創舉。
產前檢測顯著降SMA發生率
除了政府全面補助的新生兒篩檢項目外,國人熟知的唐氏症產前篩檢,就是基因檢測技術中,第一項應用於臨床且大獲成功的案例。
台基盟生技執行長黃昭熹表示,在基因檢測技術尚未完全純熟的當時,由於唐氏症只需檢測到胎兒的「第21號染色體是否多出一條」,檢測解析度相對不需太高,因此相當容易。
眼見唐氏症的成功,加上基因檢測技術、解析度不斷提高,成本亦下降中,因此,也有業者選擇投入這塊罕病基因檢測藍海,例如訊聯生技旗下的創源生技,就是國內首家投入脊髓性肌肉萎縮症(SMA)帶因檢測的業者。
創源執行長蔡政憲表示,10多年前,他因緣際會下認識了一位以色列SMA患者後,決心投入這項帶因率並不低(1/40~1/60)的罕病隱性遺傳疾病檢測。
蔡政憲表示,2005年推出SMA產前檢測後,目前為止,創源已檢測超過50萬人次,國內SMA案例也逐年降低,自2005年每年仍有50多名患者,一路下滑到近幾年只有個位數,顯見檢測防治疾病的效果。
創源目前的孕期檢測可檢測的項目,在罕病上除了SMA外,還包括X染色體脆折症篩檢(FXS),多疾病帶因檢測中,則囊括:海洋性貧血、遺傳性耳聾、裘馨氏肌肉萎縮症、龐貝氏症、苯酮尿症、威爾森氏症等。

創源執行長蔡政憲表示,10 多年前,他因緣際會下認識了一位以色列SMA 患者後,決心投入這項帶因率並不低(1/40~1/60) 的罕病隱性遺傳疾病檢測。( 攝影/ 羅翊方)
基因定序探索更多未知罕病基因
2022年12月13日,英國政府宣布將斥資超過1.75億英鎊(約2.15億美元),將英國在基因體領域的優勢落實到醫療保健系統中,其中有1.05億英鎊(約1.3億美元)將用於在全國招募10萬名新生兒進行基因體定序,幫助更早發現、治療帶有罕見遺傳疾病的新生兒。
這項消息,對國內、外針對罕病進行基因體研究的產、學界而言,可說是顆震撼彈!
亞太生醫矽谷精準醫療旗艦計畫總主持人,國衛院特聘研究員蔡世峯就直言:「英國真的把基因體科學當成關鍵國家競爭力在經營了!」
另外,蔡世峯領導的「亞太生醫矽谷精準醫療旗艦計畫」中,將罕病納入計畫研究範圍,也是國內針對更多未知罕病投入基因檢測、相關研究的重要舉措。
「該計畫透過給予罕病病人及其家人,進行完整的全基因體定序(WGS)或全外顯子定序(WES),期望解出更多目前仍未解的罕病遺傳之謎。」蔡世峯說。
在該計畫中,國衛院也進一步與罕見疾病基金會攜手成立「台灣罕病研究網絡(Taiwan Rare Disease Network, TRDN)」,串聯國內數家醫學中心,協助病患達到確診及提前預防與規劃。

國衛院特聘研究員蔡世峯領導計畫中,透過給予罕病病人及其家人,進行完整的全基因體定序(WGS) 或全外顯子定序(WES),期望解出更多目前仍未解的罕病遺傳之謎。( 攝影/ 李林璦)
FDA 對孤兒藥有哪些鼓勵措施?
美國在1983年通過孤兒藥法案(ODA),FDA所提出的主要激勵措施,包括:⑴ 提供共計超過1,800萬美元的研發補助;⑵ 藥品獲批時,高達50%的臨床開發費用可抵減稅額;⑶ 可享有7年上市後市場獨賣權;⑷ 免除超過200萬美元的處方藥使用者費用(Prescription Drug User Fee Act, PDUFA)。
此外,符合某些特定罕見、未滿足醫療需求者,FDA還祭出優先審查憑證(Priority Review Voucher);符合罕見兒科疾病者,也有罕見兒科疾病認定(RPD),皆可作為加速藥品上市的途徑。
(編按:PDUFA是美國國會於1992年通過的一項法律,該法律允許FDA從藥品製造商處收取費用,以資助新的藥品批准程序。該法案規定,FDA有權在提交新藥申請或生物製品許可證申請時,向藥品製造商收取大量申請費用,這些資金僅指定用於藥物評估和研究中心或生物製品評估中心研究藥品批准的活動。)
蔡政憲:受檢者為中心,多重檢測團結合作,才能真正預防罕病發生!
創源執行長蔡政憲指出,染色體和基因異常其實是遺傳罕病發生的一個警訊!
臺灣廠商除了致力於開發罕病療法外,在精準醫療起飛的時代,患病前的預防更加重要!
在156萬份調查中,35歲以下女性,完全正常胚胎的比例高達55%,但只要區間增加2年,35至37歲的女性,胚胎完全正常比例就會掉10%,只剩45%,41~42歲的女性,其胚胎完全正常的比例就掉到23%,只有不到1/4的機率是完全正常的胚胎,就容易不孕或者生出患有疾病的孩子。
因此,凸顯了婚前、孕前、產前基因檢測的重要性,訊聯生技旗下的創源生技,就是國內首家投入脊髓性肌肉萎縮症(SMA)帶因檢測的業者。
蔡政憲指出,SMA是全球帶因率最高的自體隱性遺傳疾病,帶因率高達1/50,創源從2005年便推出SMA產前檢測,至今檢測已超過50萬人次,也讓國內SMA案例逐年降低,近幾年新增患者僅有個位數。
「這幾來的努力、收集醫療大數據都是想要說服社會、醫師、國家資源的分配者,能看到罕病的盲點,很多疾病被冠上罕見,但以帶因率來看,其實一點都不罕見。」蔡政憲說。
蔡政憲也強調,「罕病是可以被預防、並有配套措施的!」如創源開發的胚胎染色體檢測(PGT-M)技術,可以利用胚胎在體外受精的第五天左右,抽出一部分細胞,得知哪些胚胎是健康的,如此一來,若夫妻兩人都是帶因者,可在胚胎體外進行篩檢,或是透過羊水進行產檢,就不會對生出罕病寶寶而感到恐慌。
他也表示,很多罕病的病因並不是單一一種基因突變,而是有多種複雜的致病因子與遺傳模式,因此他呼籲,基因檢測單位、羊水實驗室與醫療機構要團結合作,以受檢者為中心來進行多重檢測。
蔡政憲分享小胖威利症的一個真實案例:在受測者第一孕期時進行非侵入式產前檢測,從母血檢測中發現孩子第15號染色體可能有3條,但是還有機會在胚胎發育時進行自我修正;第二孕期繼續追蹤時,進行羊水晶片與核型分析,發現真的修正成2條染色體,但在發出正常報告前,統整數據發現,修正掉的竟是錯誤的染色體。
「因此,資訊必須圍繞受測者進行多方串聯,才能合作真正預防複雜的遺傳疾病發生。」蔡政憲強調。
台灣罕病研究網絡

台灣罕病研究網絡針對常見罕病以及其他無法確診的罕病類型進行分析,並藉此機會建立完整的網絡與資料庫。( 圖/ vecteezy)
蔡世峯表示,「台灣罕病研究網絡(Taiwan Rare Disease Network, TRDN)」 主要針對免疫缺乏、癲癇、脊髓性小腦萎縮症、聽覺障礙、雷特氏症、自體發炎疾病、以及其他無法確診的罕病類型進行分析,並藉此機會建立完整的網絡與資料庫。
截至目前為止,該團隊已完成約600個家庭基因分析。
蔡世峯表示,他們以目前國際上最標準的罕病分析方式――以一家三人組(病人及其父母,Trio)為一對進行分析,將每個人的全基因體序列,和人類基因體參考序列(Human Genome Reference Sequence)進行比對,找出每個人的基因體特徵,再運用公開的資料庫進行遺傳分析,並比對文獻,來找出可以解釋病人症狀的基因變異。
未來,他們也期望藉由這些資料了解基礎科學上致病的機轉,甚至可能進一步建立疾病模型,此外,也可持續加入長期追蹤下之病人治療後的結果,對罕病的治療、照護會更有實質上的幫助。
【附錄】臺灣孤兒藥開發公司
>>本文刊登於《環球生技月刊》Vol. 102



